Etude de la méthylation par pyroséquençage
Il s'agit d'une technologie de séquençage direct qui permet d'obtenir rapidement un profil de méthylation pour tous les doublets CG d'une région donnée. Cependant, contrairement au clonage, elle ne donne aucune indication quant aux haplotypes de méthylation (c'est à dire quant à la répartition des CG méthylés et déméthylés sur les deux allèles). Son principal avantage est sa rapidité qui autorise enfin l'analyse de grandes séries d'individus et à envisager de réaliser une cartographie des sites normalement méthylés et déméthylés dans le génome humain.
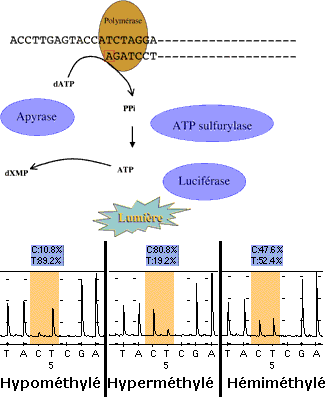
Principe du Pyroséquençage
Les nucléotides ne sont pas ajoutés tous ensemble comme dans les réactions de séquence normales, mais l'un après l'autre. Si le nucléotide ajouté dans le milieu réactionnel correspond à celui attendu par la Polymérase, il est incorporé dans le brin en cours de syntèse en libérant un PyroPhosphate. Grâce à une ATP Sulfurylase, ce pyrophosphate est transformé en ATP qui est utilisé par une luciférase pour émettre un signal lumineux. C'est ce signal qui est capté par le séquenceur qui le reproduit sous la forme d'un pic sur le Pyrogramme. La hauteur de ce pic est fonction de l'intensité du signal lumineux, elle-même proportionnelle au nombre de nucléotides incorporés en même temps. On peut donc déduire la séquence de la taille des pics obtenus. Par ailleurs, en cas de mélange de nucléotides à une même position (polymorphisme de séquence), la taille des pics permet d'avoir une quantification de la proportion de brins porteurs de l'un ou l'autre des nucléotides.
Application à l'étude de méthylation : après traitement au Bisulphite de l'ADN, l'information épigénétique est transformée en un polymorphisme génétique C ou T au niveau de chaque CG de la région. L'ADN est dans un premier temps amplifié par PCR, puis passé au Pyroséquenceur. Les porportions respectives de C et de T pour chaque position peuvent donc être mesurées par pyroséquençage, et cette proportion reflète le pourcentage de brins méthylés ou déméthylés à ce niveau dans l'échantillon initial.
On obtient donc une mesure précise et quantitative de la proportion de CG méthylés et déméthylés pour chaque position, mesure qui repose non pas sur l'analyse de quelques brins comme dans le cas du clonage, mais sur l'ensemble des molécules d'ADN présentes initialement dans l'échantillon. Dans la mesure ou cette analyse est rapide, on peut envisager de passer aisément un grand nombre d'échantillons. Il existe cependant des limites : la taille des fragments analysables ne dépasse pas en moyenne 50 - 60 nucléotides à chaque séquence et cette technique ne donne aucune information quant à la répartition de la méthylation sur les deux allèles. Par exemple, si une position est méthylée à 50%, cela peut correspondre soit à un allèle toujours méthylé et l'autre toujours déméthylé (cas des DMRs), soit à une position méthylée dans 50 % des cas sans spécificité d'allèle.
![]()